
Principal component analysis (PCA) is routinely employed on a wide range of problems. From the detection of outliers to predictive modeling, PCA has the ability of projecting the observations described by 


It is an unsupervised method, meaning it will always look into the greatest sources of variation regardless of the data structure. Its counterpart, the partial least squares (PLS), is a supervised method and will perform the same sort of covariance decomposition, albeit building a user-defined number of components (frequently designated as latent variables) that minimize the SSE from predicting a specified outcome with an ordinary least squares (OLS). The PLS is worth an entire post and so I will refrain from casting a second spotlight.
In case PCA is entirely new to you, there is an excellent Primer from Nature Biotechnology that I highly recommend. Notwithstanding the focus on life sciences, it should still be clear to others than biologists.
Mathematical foundation
There are numerous PCA formulations in the literature dating back as long as one century, but all in all PCA is pure linear algebra. One of the most popular methods is the singular value decomposition (SVD). The SVD algorithm breaks down a matrix 
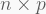

where 


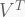
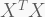

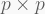

PCA reduces the 


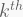
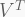
- PCs are ordered by the decreasing amount of variance explained
- PCs are orthogonal i.e. uncorrelated to each other
- The columns of
- SVD-based PCA does not tolerate missing values (but there are solutions we will cover shortly)

For a more elaborate explanation with introductory linear algebra, here is an excellent free SVD tutorial I found online. At any rate, I guarantee you can master PCA without fully understanding the process.
Let’s get started with R
Although there is a plethora of PCA methods available for R, I will only introduce two,
- prcomp, a default function from the R base package
- pcaMethods, a Bioconductor package that I frequently use for my own PCAs
I will start by demonstrating that prcomp is based on the SVD algorithm, using the base svd function.
| # Generate scaled 4*5 matrix with random std normal samples | |
| set.seed(101) | |
| mat <- scale(matrix(rnorm(20), 4, 5)) | |
| dimnames(mat) <- list(paste("Sample", 1:4), paste("Var", 1:5)) | |
| # Perform PCA | |
| myPCA <- prcomp(mat, scale. = F, center = F) | |
| myPCA$rotation # loadings | |
| myPCA$x # scores |
By default, prcomp will retrieve 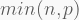
| # Perform SVD | |
| mySVD <- svd(mat) | |
| mySVD # the diagonal of Sigma mySVD$d is given as a vector | |
| sigma <- matrix(0,4,4) # we have 4 PCs, no need for a 5th column | |
| diag(sigma) <- mySVD$d # sigma is now our true sigma matrix |
Now that we have the PCA and SVD objects, let us compare the respective scores and loadings. We will compare the scores from the PCA with the product of 
| # Compare PCA scores with the SVD's U*Sigma | |
| theoreticalScores <- mySVD$u %*% sigma | |
| all(round(myPCA$x,5) == round(theoreticalScores,5)) # TRUE | |
| # Compare PCA loadings with the SVD's V | |
| all(round(myPCA$rotation,5) == round(mySVD$v,5)) # TRUE | |
| # Show that mat == U*Sigma*t(V) | |
| recoverMatSVD <- theoreticalScores %*% t(mySVD$v) | |
| all(round(mat,5) == round(recoverMatSVD,5)) # TRUE | |
| # Show that mat == scores*t(loadings) | |
| recoverMatPCA <- myPCA$x %*% t(myPCA$rotation) | |
| all(round(mat,5) == round(recoverMatPCA,5)) # TRUE |
PCA of the wine data set
Now that we established the association between SVD and PCA, we will perform PCA on real data. I found a wine data set at the UCI Machine Learning Repository that might serve as a good starting example.
| wine <- read.table("http://archive.ics.uci.edu/ml/machine-learning-databases/wine/wine.data", | |
| sep=",") |
According to the documentation, these data consist of 13 physicochemical parameters measured in 178 wine samples from three distinct cultivars grown in Italy. Let’s check patterns in pairs of variables, and then see what a PCA does about that by plotting PC1 against PC2.
| # Name the variables | |
| colnames(wine) <- c("Cvs","Alcohol","Malic acid","Ash", | |
| "Alcalinity of ash", "Magnesium", | |
| "Total phenols", "Flavanoids", | |
| "Nonflavanoid phenols", "Proanthocyanins", | |
| "Color intensity", "Hue", | |
| "OD280/OD315 of diluted wines", "Proline") | |
| # The first column corresponds to the classes | |
| wineClasses <- factor(wine$Cvs) | |
| # Use pairs | |
| pairs(wine[,-1], col = wineClasses, upper.panel = NULL, | |
| pch = 16, cex = 0.5) | |
| legend("topright", bty = "n", legend = c("Cv1","Cv2","Cv3"), | |
| pch = 16, col = c("black","red","green"), | |
| xpd = T, cex = 2, y.intersp = 0.5) |

Among other things, we observe correlations between variables (e.g. total phenols and flavonoids), and occasionally the two-dimensional separation of the three cultivars (e.g. using alcohol % and the OD ratio).
If its hard enough looking into all pairwise interactions in a set of 13 variables, let alone in sets of hundreds or thousands of variables. In these instances PCA is of great help. Let’s give it a try in this data set:
| dev.off() # clear the format from the previous plot | |
| winePCA <- prcomp(scale(wine[,-1])) | |
| plot(winePCA$x[,1:2], col = wineClasses) |

Three lines of code and we see a clear separation among grape vine cultivars. In addition, the data points are evenly scattered over relatively narrow ranges in both PCs. We could next investigate which parameters contribute the most to this separation and how much variance is explained by each PC, but I will leave it for pcaMethods. We will now repeat the procedure after introducing an outlier in place of the 10th observation.
| wineOutlier <- wine | |
| wineOutlier[10,] <- wineOutlier[10,]*10 # change the 10th obs. into an extreme one by multiplying its profile by 10 | |
| outlierPCA <- prcomp(scale(wineOutlier[,-1])) | |
| plot(outlierPCA$x[,1:2], col = wineClasses) |

As expected, the huge variance stemming from the separation of the 10th observation from the core of all other samples is fully absorbed by the first PC. The outlying sample becomes plain evident.
PCA of the wine data set with pcaMethods
We will now turn to pcaMethods, a compact suite of PCA tools. First you will need to install it from the Bioconductor:
| if (!requireNamespace("BiocManager", quietly = TRUE)) | |
| install.packages("BiocManager") | |
| BiocManager::install("pcaMethods") | |
| library(pcaMethods) |
There are three mains reasons why I use pcaMethods so extensively:
- Besides SVD, it provides several different methods (bayesian PCA, probabilistic PCA, robust PCA, to name a few)
- Some of these algorithms tolerate and impute missing values
- The object structure and plotting capabilities are user-friendly
All information available about the package can be found here. I will now simply show the joint scores-loadings plots, but still encourage you to explore it further.
I will select the default SVD method to reproduce our previous PCA result, with the same scaling strategy as before (UV, or unit-variance, as executed by scale). The argument scoresLoadings gives you control over printing scores, loadings, or both jointly as right next. The standard graphical parameters (e.g. cex, pch, col) preceded by either letters s or l control the aesthetics in the scores or loadings plots, respectively.
| winePCAmethods <- pca(wine[,-1], scale = "uv", center = T, | |
| nPcs = 2, method = "svd") | |
| slplot(winePCAmethods, scoresLoadings = c(T,T), | |
| scol = wineClasses) |
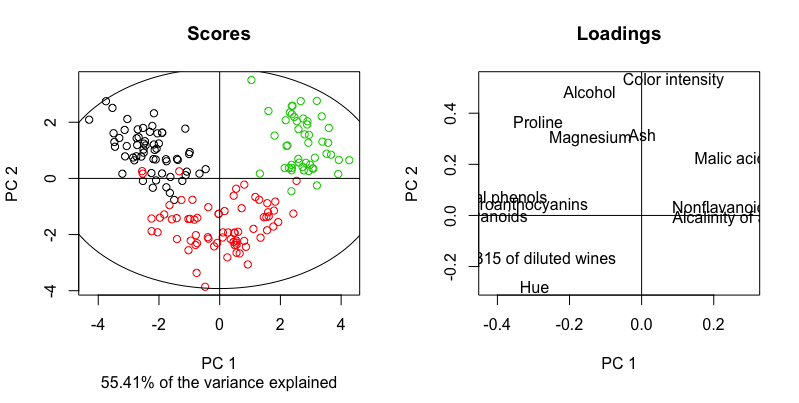
So firstly, we have a faithful reproduction of the previous PCA plot. Then, having the loadings panel on its right side, we can claim that
- Wine from Cv2 (red) has a lighter color intensity, lower alcohol %, a greater OD ratio and hue, compared to the wine from Cv1 and Cv3.
- Wine from Cv3 (green) has a higher content of malic acid and non-flavanoid phenols, and a higher alkalinity of ash compared to the wine from Cv1 (black)
Finally, although the variance jointly explained by the first two PCs is printed by default (55.41%), it might be more informative consulting the variance explained in individual PCs. We can call the structure of winePCAmethods, inspect the slots and print those of interest, since there is a lot of information contained. The variance explained per component is stored in a slot named R2.
| str(winePCAmethods) # slots are marked with @ | |
| winePCAmethods@R2 |
Seemingly, PC1 and PC2 explain 36.2% and 19.2% of the variance in the wine data set, respectively. PCAs of data exhibiting strong effects (such as the outlier example given above) will likely result in the sequence of PCs showing an abrupt drop in the variance explained. Screeplots are helpful in that matter, and allow you determining how much variance you can put into a principal component regression (PCR), for example, which is exactly what we will try next.
PCR with the housing data set
Now we will tackle a regression problem using PCR. I will use an old housing data set also deposited in the UCI MLR. Again according to its documentation, these data consist of 14 variables and 504 records from distinct towns somewhere in the US. To perform PCR all we need is conduct PCA and feed the scores of 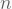
| houses <- read.table("http://archive.ics.uci.edu/ml/machine-learning-databases/housing/housing.data", | |
| header = F, na.string = "?") | |
| colnames(houses) <- c("CRIM", "ZN", "INDUS","CHAS", | |
| "NOX","RM","AGE","DIS","RAD", | |
| "TAX","PTRATIO","B","LSTAT","MEDV") | |
| # Perform PCA | |
| pcaHouses <- prcomp(scale(houses[,-14])) | |
| scoresHouses <- pcaHouses$x | |
| # Fit lm using the first 3 PCs | |
| modHouses <- lm(houses$MEDV ~ scoresHouses[,1:3]) | |
| summary(modHouses) |
The printed summary shows two important pieces of information. Firstly, the three estimated coefficients (plus the intercept) are considered significant (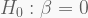

Next we will compare this simple model to a OLS model featuring all 14 variables, and finally compare the observed vs. predicted MEDV plots from both models. Note that in the lm syntax, the response 

| # Fit lm using all 14 vars | |
| modHousesFull <- lm(MEDV ~ ., data = houses) | |
| summary(modHousesFull) # R2 = 0.741 | |
| # Compare obs. vs. pred. plots | |
| par(mfrow = c(1,2)) | |
| plot(houses$MEDV, predict(modHouses), | |
| xlab = "Observed MEDV", ylab = "Predicted MEDV", | |
| main = "PCR", abline(a = 0, b = 1, col = "red")) | |
| plot(houses$MEDV, predict(modHousesFull), | |
| xlab = "Observed MEDV", ylab = "Predicted MEDV", | |
| main = "Full model", abline(a = 0, b = 1, col = "red")) |

Here the full model displays a slight improvement in fit (
Just as a side note, you probably noticed both models underestimated the MEDV in towns with MEVD worth 50,000 dollars. My guess is that missing values were set to MEVD = 50.
Concluding,
- The SVD algorithm is founded on fundamental properties of linear algebra including matrix diagonalization. SVD-based PCA takes part of its solution and retains a reduced number of orthogonal covariates that explain as much variance as possible.
- Use PCA when handling high-dimensional data. It is insensitive to correlation among variables and efficient in detecting sample outliers.
- If you plan to use PCA results for subsequent analyses all care should be undertaken in the process. Although typically outperformed by numerous methods, PCR still benefits from interpretability and can be effective in many settings.
All feedback from these tutorials is very welcome, please enter the Contact tab and leave your comments. I do also appreciate suggestions. Enjoy!

nicely explained..well done sir!
LikeLike
Good job please treat PCA using mintab to solve a problem and kindkly explain in details kore than you do for R. Thanks
LikeLike
Hi Owolabi. I presume you meant “minitab”? I apologize but the focus of my blog is R. I hope you can find the information you seek somewhere else! Best regards
LikeLike
Dear Francisco, thank you for your blogs, it is a bit of “auto promotion” but if you are interested in PCA based methods you could also look at FactoMineR http://factominer.free.fr/ and there are many videos https://www.youtube.com/playlist?list=PLnZgp6epRBbTsZEFXi_p6W48HhNyqwxIu
Best wishes,
Julie
LikeLike
Did you make the animation at the start of the article with R? I had some idea about PCA, but even before going through the article, the animation gave me so much more insight. Thanks for a great job!
LikeLike
This is an awesome explanation, but SO many variables. I only have two. How would I do this with only two variables?
LikeLike
Hi Alterra, thank you! The same as with three or more. The animation on top illustrates the two-dimensional case very well. Is your question how to do it mathematically, via the SVD algorithm?
Francisco
LikeLike
Hi Francisco, thank you very much for this post! I found it very helpful and enjoyable to read (also the references to the PCA and SVD resources were great).
As I followed your analysis, I tried to install the library pcaMethods and I found that the installation is different for the new versions of R.
The following code worked for me:
if (!requireNamespace(“BiocManager”, quietly = TRUE))
+ install.packages(“BiocManager”)
BiocManager::install(version = “3.11”)
BiocManager::install(c(“pcaMethods”))
Maybe this can help others.
LikeLike
Hi Fede,
Thanks for your feedback! I might follow your code example to correct mine above, greetings.
Francisco
LikeLike
Hi Francisco
I’ve been struggling all weekend because on Tuesday I want to teach my Analytical Chemistry students how to do Principal Component Analysis in R. I have been using R since this May, and I have been studying PCA on my own for six weeks. I have been watching many videos and reading many tutorials. Yours was able to make it clear for me, and in the light of it I am going to revisit the other materials and try to discover what I was missing.
I appreciate how I was able to copy your code and paste it into R and have it run.
Thank you!
LikeLike
Dear Greg, thanks for the kind words. I am glad you found it useful, hopefully your students will build an intuition around PCA and its applications. Greetings and have a nice week, Francisco
LikeLike
Question for you Francisco
I wan students to be able to copy and paste commands from my notes. That’s not working when I use google docs–I’m getting errors when I paste a command, but it works when I retype it in R. What did you use to display R commands in your tutorial?
LikeLike
Hi Greg, I suggest using a code-syntax friendly text editor, you have plenty of free options – Notepad++, Sublime or Atom to name a few. The best option in my mind is having your students installing RStudio and pasting the code into a new, blank script. Greetings
LikeLike