

Statistical models generally assume that
- All observations are independent from each other
- The distribution of the residuals follows
, irrespective of the values taken by the dependent variable y
When any of the two is not observed, more sophisticated modelling approaches are necessary. Let’s consider two hypothetical problems that violate the two respective assumptions, where y denotes the dependent variable:
A. Suppose you want to study the relationship between average income (y) and the educational level in the population of a town comprising four fully segregated blocks. You will sample 1,000 individuals irrespective of their blocks. If you model as such, you neglect dependencies among observations – individuals from the same block are not independent, yielding residuals that correlate within block.
B. Suppose you want to study the relationship between anxiety (y) and the levels of triglycerides and uric acid in blood samples from 1,000 people, measured 10 times in the course of 24 hours. If you model as such, you will likely find that the variance of y changes over time – this is an example of heteroscedasticity, a phenomenon characterized by the heterogeneity in the variance of the residuals.
In A. we have a problem of dependency caused by spatial correlation, whereas in B. we have a problem of heterogeneous variance. As a result, classic linear models cannot help in these hypothetical problems, but both can be addressed using linear mixed-effect models (LMMs). In rigour though, you do not need LMMs to address the second problem.
LMMs are extraordinarily powerful, yet their complexity undermines the appreciation from a broader community. LMMs dissect hierarchical and / or longitudinal (i.e. time course) data by separating the variance due to random sampling from the main effects. In essence, on top of the fixed effects normally used in classic linear models, LMMs resolve i) correlated residuals by introducing random effects that account for differences among random samples, and ii) heterogeneous variance using specific variance functions, thereby improving the estimation accuracy and interpretation of fixed effects in one go.
I personally reckon that most relevant textbooks and papers are hard to grasp for non-mathematicians. Therefore, following the brief reference in my last post on GWAS I will dedicate the present tutorial to LMMs. For further reading I highly recommend the ecology-oriented Zuur et al. (2009) and the R-intensive Gałecki et al. (2013) books, and this simple tutorial from Bodo Winter. For agronomic applications, H.-P. Piepho et al. (2003) is an excellent theoretical introduction.
Here, we will build LMMs using the Arabidopsis dataset from the package lme4, from a study published by Banta et al. (2010). These data summarize variation in total fruit set per plant in Arabidopsis thaliana plants conditioned to fertilization and simulated herbivory. Our goal is to understand the effect of fertilization and simulated herbivory adjusted to experimental differences across groups of plants.
Mixed-effect linear models
Whereas the classic linear model with n observational units and p predictors has the vectorized form

with the 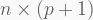
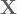

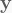

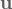

where 
One of the most common doubts concerning LMMs is determining whether a variable is a random or fixed. First of all, an effect might be fixed, random or even both simultaneously – it largely depends on how you approach a given problem. Generally, you should consider all factors that qualify as sampling from a population as random effects (e.g. individuals in repeated measurements, cities within countries, field trials, plots, blocks, batches) and everything else as fixed. As a rule of thumb, i) factors with fewer than 5 levels should be considered fixed and conversely ii) factors with numerous levels should be considered random effects in order to increase the accuracy in the estimation of variance. Both points relate to the LMM assumption of having normally distributed random effects.
Best linear unbiased estimators (BLUEs) and predictors (BLUPs) correspond to the values of fixed and random effects, respectively. The usage of the so-called genomic BLUPs (GBLUPs), for instance, elucidates the genetic merit of animal or plant genotypes that are regarded as random effects when trial conditions, e.g. location and year of trials are considered fixed. However, many studies sought the opposite, i.e. using breeding values as fixed effects and trial conditions as random, when the levels of the latter outnumber the former, chiefly because of point ii) outlined above. In GWAS, LMMs aid in teasing out population structure from the phenotypic measures.
Let’s get started with R
We will follow a structure similar to the 10-step protocol outlined in Zuur et al. (2009): i) fit a full ordinary least squares model and run the diagnostics in order to understand if and what is faulty about its fit; ii) fit an identical generalized linear model (GLM) estimated with ML, to serve as a reference for subsequent LMMs; iii) deploy the first LMM by introducing random effects and compare to the GLM, optimize the random structure in subsequent LMMs; iv) optimize the fixed structure by determining the significant of fixed effects, always using ML estimation; finally, v) use REML estimation on the optimal model and interpret the results.
You need to havenlme andlme4 installed to proceed. We will firstly examine the structure of the Arabidopsis dataset.
| # Install (if necessary) and load nlme and lme4 | |
| library(nlme) | |
| library(lme4) | |
| # Load dataset, inspect size and additional info | |
| data(Arabidopsis) | |
| dim(Arabidopsis) # 625 observations, 8 variables | |
| ?Arabidopsis | |
| attach(Arabidopsis) |
The Arabidopsis dataset describes 625 plants with respect to the the following 8 variables (transcript from R):
reg- region: a factor with 3 levels
NL(Netherlands),SP(Spain),SW(Sweden) popu- population: a factor with the form
n.Rrepresenting a population in regionR gen- genotype: a factor with 24 (numeric-valued) levels
rack- a nuisance factor with 2 levels, one for each of two greenhouse racks
nutrient- fertilization treatment/nutrient level (1, minimal nutrients or 8, added nutrients)
amd- simulated herbivory or “clipping” (apical meristem damage):
unclipped(baseline) orclipped status- a nuisance factor for germination method (
Normal,Petri.Plate, orTransplant) total.fruits- total fruit set per plant (integer), henceforth TFPP for short.
We will now visualise the absolute frequencies in all 7 factors and the distribution for TFPP.
| # Overview of the variables | |
| par(mfrow = c(2,4)) | |
| barplot(table(reg), ylab = "Frequency", main = "Region") | |
| barplot(table(popu), ylab = "Frequency", main = "Population") | |
| barplot(table(gen), ylab = "Frequency", las = 2, main = "Genotype") | |
| barplot(table(rack), ylab = "Frequency", main = "Rack") | |
| barplot(table(nutrient), ylab = "Frequency", main = "Nutrient") | |
| barplot(table(amd), ylab = "Frequency", main = "AMD") | |
| barplot(table(status), ylab = "Frequency", main = "Status") | |
| hist(total.fruits, col = "grey", main = "Total fruits", xlab = NULL) |
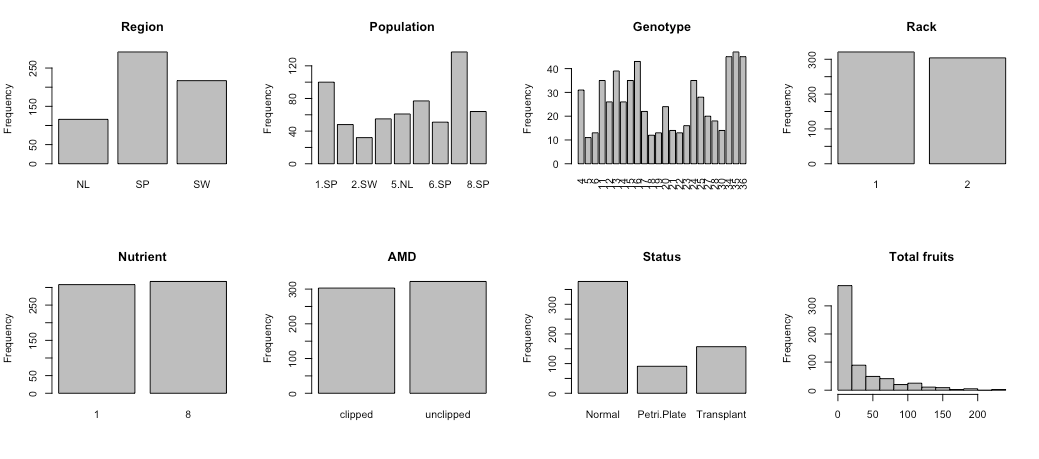
The frequencies are overall balanced, perhaps except for status (i.e. germination method). Genotype, greenhouse rack and fertilizer are incorrectly interpreted as quantitative variables. In addition, the distribution of TFPP is right-skewed. As such, we will encode these three variables as categorical variables and log-transform TFPP to approximate a Gaussian distribution (natural logarithm).
| # Transform the three factor variables gen, rack and nutrient | |
| Arabidopsis[,c("gen","rack","nutrient")] <- lapply(Arabidopsis[,c("gen","rack","nutrient")], factor) | |
| str(Arabidopsis) | |
| # Re-attach after correction, ignore warnings | |
| attach(Arabidopsis) | |
| # Add 1 to total fruits, otherwise log of 0 will prompt error | |
| total.fruits <- log(1 + total.fruits) |
A closer look into the variables shows that each genotype is exclusive to a single region. The data contain no missing values.
| # gen x popu table | |
| table(gen, popu) | |
| # Any NAs? | |
| any(is.na(Arabidopsis)) # FALSE |
Formula syntax basics
At this point I hope you are familiar with the formula syntax in R. Note that interaction terms are denoted by : and fully crossed effects with *, so that A*B = A + B + A:B. The following code example
lm(y ~ x1 + x2*x3)
builds a linear model of y using 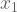

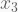
I. You can also introduce polynomial terms with the function poly. One handy trick I use to expand all pairwise interactions among predictors is
model.matrix(y ~ .*., data = X)
provided a matrix X that gathers all predictors and y. You can also simply use .*. inside the lm call, however you will likely need to preprocess the resulting interaction terms.
While the syntax of lme is identical to lm for fixed effects, its random effects are specified under the argument random as
random = ~intercept + fixed effect | random effect
and can be nested using /. In the following example
random = ~1 + C | A/B
the random effect B is nested within random effect A, altogether with random intercept and slope with respect to C. Therefore, not only will the groups defined by A and A/B have different intercepts, they will also be explained by different slight shifts of
Classic linear model
Ideally, you should start will a full model (i.e. including all independent variables). Here, however, we cannot use all descriptors in the classic linear model since the fit will be singular due to the redundancy in the levels of reg and popu. For simplicity I will exclude these alongside gen, since it contains a lot of levels and also represents a random sample (from many other extant Arabidopsis genotypes). Additionally, I would rather use rack and status as random effects in the following models but note that having only two and three levels respectively, it is advisable to keep them as fixed.
| LM <- lm(total.fruits ~ rack + nutrient + amd + status) | |
| summary(LM) | |
| par(mfrow = c(2,2)) | |
| plot(LM) |
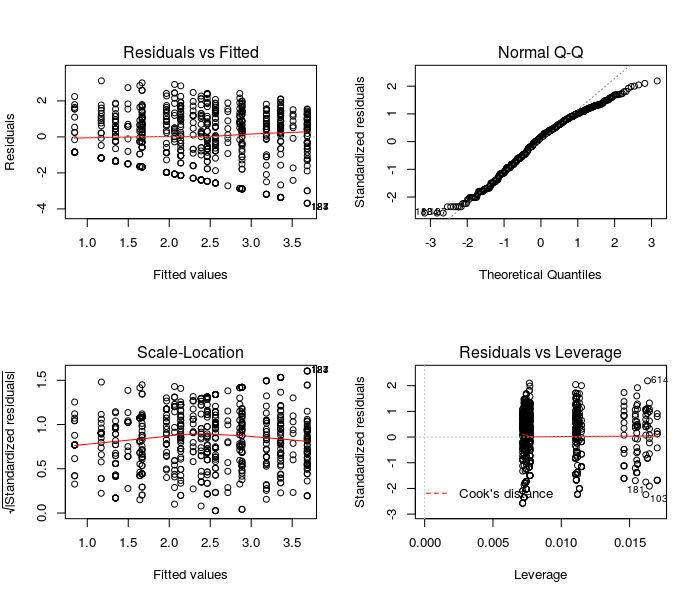
These diagnostic plots show that the residuals of the classic linear model poorly qualify as normally distributed. Because we have no obvious outliers, the leverage analysis provides acceptable results. We will try to improve the distribution of the residuals using LMMs.
Generalized linear model
We need to build a GLM as a benchmark for the subsequent LMMs. This model can be fit without random effects, just like a lm but employing ML or REML estimation, using the gls function. Hence, it can be used as a proper null model with respect to random effects. The GLM is also sufficient to tackle heterogeneous variance in the residuals by leveraging different types of variance and correlation functions, when no random effects are present (see arguments correlation and weights).
| GLM <- gls(total.fruits ~ rack + nutrient + amd + status, | |
| method = "ML") | |
| summary(GLM) |
At this point you might consider comparing the GLM and the classic linear model and note they are identical. Also, you might wonder why are we using LM instead of REML – as hinted in the introduction, REML comparisons are meaningless in LMMs that differ in their fixed effects. Therefore, we will base all of our comparisons on LM and only use the REML estimation on the final, optimal model.
Optimal random structure
Let’s fit our first LMM with all fixed effects used in the GLM and introducing reg, popu, gen, reg/popu, reg/gen, popu/gen and reg/popu/gen as random intercepts, separately.
In order to compare LMMs (and GLM), we can use the function anova (note that it does not work for lmer objects) to compute the likelihood ratio test (LRT). This test will determine if the models are significantly different with respect to goodness-of-fit, as weighted by the trade-off between variance explained and degrees-of-freedom. The model fits are also evaluated based on the Akaike (AIC) and Bayesian information criteria (BIC) – the smaller their value, the better the fit.
| lmm1 <- lme(total.fruits ~ rack + nutrient + amd + status, | |
| random = ~1|reg, method = "ML") | |
| lmm2 <- lme(total.fruits ~ rack + nutrient + amd + status, | |
| random = ~1|popu, method = "ML") | |
| lmm3 <- lme(total.fruits ~ rack + nutrient + amd + status, | |
| random = ~1|gen, method = "ML") | |
| lmm4 <- lme(total.fruits ~ rack + nutrient + amd + status, | |
| random = ~1|reg/popu, method = "ML") | |
| lmm5 <- lme(total.fruits ~ rack + nutrient + amd + status, | |
| random = ~1|reg/gen, method = "ML") | |
| lmm6 <- lme(total.fruits ~ rack + nutrient + amd + status, | |
| random = ~1|popu/gen, method = "ML") | |
| lmm7 <- lme(total.fruits ~ rack + nutrient + amd + status, | |
| random = ~1|reg/popu/gen, method = "ML") | |
| anova(GLM, lmm1, lmm2, lmm3, lmm4, lmm5, lmm6, lmm7) |
We could now base our selection on the AIC, BIC or log-likelihood. Considering most models are undistinguishable with respect to the goodness-of-fit, I will select lmm6 and lmm7 as the two best models so that we have more of a random structure to look at. Take a look into the distribution of the random effects with plot(ranef(MODEL)). We next proceed to incorporate random slopes.
There is the possibility that the different researchers from the different regions might have handled and fertilized plants differently, thereby exerting slightly different impacts. Let’s update lmm6 and lmm7 to include random slopes with respect to nutrient. We first need to setup a control setting that ensures the new models converge.
| # Set optimization pars | |
| ctrl <- lmeControl(opt="optim") | |
| lmm6.2 <- update(lmm6, .~., random = ~nutrient|popu/gen, control = ctrl) | |
| lmm7.2 <- update(lmm7, .~., random = ~nutrient|reg/popu/gen, control = ctrl) | |
| anova(lmm6, lmm6.2, lmm7, lmm7.2) # both models improved | |
| anova(lmm6.2, lmm7.2) # similar fit; lmm6.2 more parsimonious | |
| summary(lmm6.2) |
Assuming a level of significance 
nutrient improved both lmm6 and lmm7. Comparing lmm6.2 andlmm7.2 head-to-head provides no evidence for differences in fit, so we select the simpler model,lmm6.2. Let’s check how the random intercepts and slopes distribute in the highest level (i.e. gen within popu).
| plot(ranef(lmm6.2, level = 2)) |
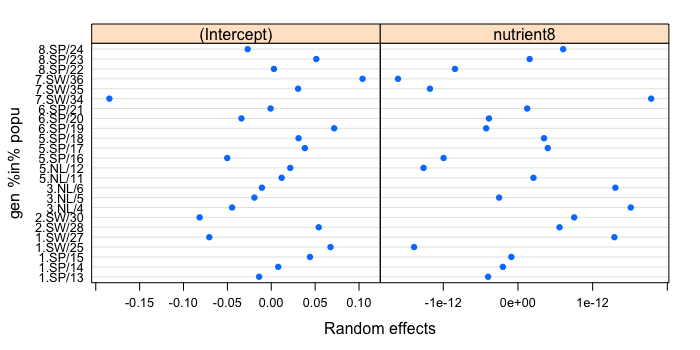
The random intercepts (left) appear to be normally distributed, except for genotype 34, biased towards negative values. This could warrant repeating the entire analysis without this genotype. The random slopes (right) appear to be normally distributed as well. Interestingly, there is a negative correlation of -0.61 between random intercepts and slopes, suggesting that genotypes with low baseline TFPP tend to respond better to fertilization. Try plot(ranef(lmm6.2, level = 1)) to observe the distributions at the level of popu only. Next, we will use QQ plots to compare the residual distributions between the GLM and lmm6.2 to gauge the relevance of the random effects.
| # QQ plots (drawn to the same scale!) | |
| par(mfrow = c(1,2)) | |
| lims <- c(-3.5,3.5) | |
| qqnorm(resid(GLM, type = "normalized"), | |
| xlim = lims, ylim = lims,main = "GLM") | |
| abline(0,1, col = "red", lty = 2) | |
| qqnorm(resid(lmm6.2, type = "normalized"), | |
| xlim = lims, ylim = lims, main = "lmm6.2") | |
| abline(0,1, col = "red", lty = 2) |
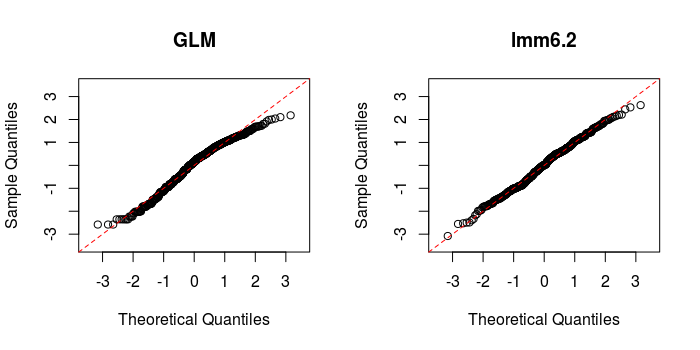
The improvement is clear. Bear in mind these results do not change with REML estimation. Try different arrangements of random effects with nesting and random slopes, explore as much as possible!
Optimal fixed structure
Now that we are happy with the random structure, we will look into the summary of the optimal model so far (i.e. lmm6.2) and determine if we need to modify the fixed structure.
| summary(lmm6.2) |
All effects are significant with status that represents transplanted plants. Given the significant effect from the other two levels, we will keep status and all current fixed effects. Just for fun, let’s add the interaction term nutrient:amd and see if there is any significant improvement in fit.
| lmm8 <- update(lmm6.2, .~. + nutrient:amd) | |
| summary(lmm8) | |
| anova(lmm8, lmm6.2) |
The addition of the interaction was non-significant with respect to both
Fit optimal model with REML
We could play a lot more with different model structures, but to keep it simple let’s finalize the analysis by fitting the lmm6.2 model using REML and finally identifying and understanding the differences in the main effects caused by the introduction of random effects.
| finalModel <- update(lmm6.2, .~., method = "REML") | |
| summary(finalModel) |
We will now contrast our REML-fitted final model against a REML-fitted GLM and determine the impact of incorporating random intercept and slope, with respect to nutrient, at the level of popu/gen. Therefore, both will be given the same fixed effects and estimated using REML.
| dev.off() # Reset previous graphical pars | |
| # New GLM, updated from the first by estimating with REML | |
| GLM2 <- update(GLM, .~., method = "REML") | |
| # Plot side by side, beta with respective SEs | |
| plot(coef(GLM2), xlab = "Fixed Effects", ylab = expression(beta), axes = F, | |
| pch = 16, col = "black", ylim = c(-.9,2.2)) | |
| stdErrors <- coef(summary(GLM2))[,2] | |
| segments(x0 = 1:6, x1 = 1:6, y0 = coef(GLM2) - stdErrors, y1 = coef(GLM2) + stdErrors, | |
| col = "black") | |
| axis(2) | |
| abline(h = 0, col = "grey", lty = 2) | |
| axis(1, at = 1:6, | |
| labels = c("Intercept", "Rack", "Nutrient (Treated)","AMD (Unclipped)","Status (PP)", | |
| "Status (Transplant)"), cex.axis = .7) | |
| # LMM | |
| points(1:6 + .1, fixef(finalModel), pch = 16, col = "red") | |
| stdErrorsLMM <- coef(summary(finalModel))[,2] | |
| segments(x0 = 1:6 + .1, x1 = 1:6 + .1, y0 = fixef(finalModel) - stdErrorsLMM, | |
| y1 = fixef(finalModel) + stdErrorsLMM, col = "red") | |
| # Legend | |
| legend("topright", legend = c("GLM","LMM"), text.col = c("black","red"), bty = "n") |
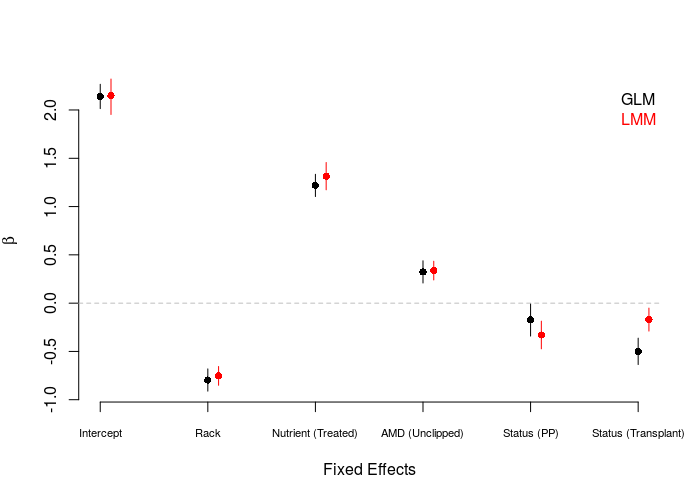
The figure above depicts the estimated nutrient, the SE is smaller in the LMM. Second, the relative effects from two levels of status are opposite. With the consideration of random effects, the LMM estimated a more negative effect of culturing in Petri plates on TFPP, and conversely a less negative effect of transplantation.
| plot(finalModel) |
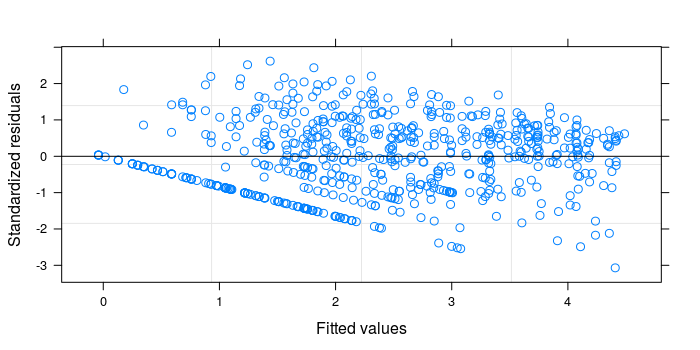
The distribution of the residuals as a function of the predicted TFPP values in the LMM is still similar to the first panel in the diagnostic plots of the classic linear model. The usage of additional predictors and generalized additive models would likely improve it.
Conclusions
Now that we account for genotype-within-region random effects, how do we interpret the LMM results?
Plants that were placed in the first rack, left unfertilized, clipped and grown normally have an average TFPP of 2.15. This is the value of the estimated grand mean (i.e. intercept), and the predicted TFPP when all other factors and levels do not apply. For example, a plant grown under the same conditions but placed in the second rack will be predicted to have a smaller yield, more precisely of 
- Fertilized plants produce more fruits than those kept unfertilized. This was the strongest main effect and represents a very sensible finding.
- Plants grown in the second rack produce less fruits than those in the first rack. This was the second strongest main effect identified. Could this be due to light / water availability?
- Simulated herbivory (AMD) negatively affects fruit yield. This is also a sensible finding – when plants are attacked, more energy is allocated to build up biochemical defence mechanisms against herbivores and pathogens, hence compromising growth and eventually fruit yield.
- Both culturing in Petri plates and transplantation, albeit indistinguishable, negatively affect fruit yield as opposed to normal growth. When conditions are radically changed, plants must adapt swiftly and this comes at a cost as well. Thus, these observations too make perfect sense.
- One important observation is that the genetic contribution to fruit yield, as gauged by
gen, was normally distributed and adequately modelled as random. One single outlier could eventually be excluded from the analysis. This makes sense assuming plants have evolved universal mechanisms in response to both nutritional status and herbivory that overrule any minor genetic differences among individuals from the same species.
Wrap-up
- Always check the residuals and the random effects! While both linear models and LMMs require normally distributed residuals with homogeneous variance, the former assumes independence among observations and the latter normally distributed random effects. Use normalized residuals to establish comparisons.
- One key additional advantage of LMMs we did not discuss is that they can handle missing values.
- Wide format data should be first converted to long format, using e.g. the R package reshape.
- Variograms are very helpful in determining spatial or temporal dependence in the residuals. In the case of spatial dependence, bubble plots nicely represent residuals in the space the observations were drown from (e.g. latitude and longitude; refer to Zuur et al. (2009) for more information).
- REML estimation is unbiased but does not allow for comparing models with different fixed structures. Only use the REML estimation on the optimal model.
With respect to this particular set of results:
- The analysis outlined here is not as exhaustive as it should be. Among other things, we did neither initially consider interaction terms among fixed effects nor investigate in sufficient depth the random effects from the optimal model.
- The dependent variable (total fruit set per plant) was highly right-skewed and required a log-transformation for basic modeling. The large amount of zeros would in rigour require zero inflated GLMs or similar approaches.
- All predictors used in the analysis were categorical factors. We could similarly use an ANOVA model. LMMs are likely more relevant in the presence of quantitative or mixed types of predictors.
I would like to thank Hans-Peter Piepho for answering my nagging questions over ResearchGate. I hope these superficial considerations were clear and insightful. I look forward for your suggestions and feedback. My next post will cover a joint multivariate model of multiple responses, the graph-guided fused LASSO (GFLASSO) using a R package I am currently developing. Happy holidays!
Citation
de Abreu e Lima, F (2017). poissonisfish: Linear mixed-effect models in R. From https://poissonisfish.com/2017/12/11/linear-mixed-effect-models-in-r/

I went over this site and I conceive you have a lot of great info, saved to bookmarks (:.
LikeLike
Thanks!
LikeLike
Dear Francisco,
I am analyzing agricultural trial data pooled from different locations (same treatment list over different sites). I was advised to build my data model as below when pooling same treatment response (e.g., yield) from different site:
SOV Df
Location l-1
Rep(Loc) l(r-1)
Treatment t-1
Location*Treatment (l-1)(t-1)
Error l(r-1)(t-1)
Total lrt-1
To do the mean separation test I was told to calculate the LSD using df and MSE from the interaction term (Location*Treatment).
I wonder does anyone here know the R code to develop the mix model above and run the LSD test using interaction term df and MSE to my personal email wangtao.home@gmail.com?
Many thanks!!
LikeLike
Hi Tao,
Apologies but I did not understand your query, if you clarify it I can eventually provide some comments! Regards,
Francisco
LikeLike
I’ve gone through this and am not seeing the negative correlation (-0.61) between random intercept and slope. Am I missing something?
LikeLike
Hi Zach, do you get some value close to -0.61? This could be a precision problem, potentially owing to new package releases. Do the plots look the same or closely identical?
LikeLike
Hi Francisco,
Fantastic article so far and has really helped me to develop my LMM. I’m wondering if you could clarify something for me however:
You noted that the data should be in Long Format for this. Can you confirm what stage of the procedure outlined this is required for? (i.e., can I perform regular LMs using long data too, or should it only be transformed from wide to long when you are ready to look at the LMMs?)
Thanks in advance,
Tom
LikeLike
Hi Tom, thanks for the kind words! I would perhaps note that the need for melting or reshaping wide to long format varies case by case – here this was not necessary. For the sake of argument, think about a time series listing timepoints as separate columns; this is a case where you would need to stack the values (e.g. seed size) in a single column and create a new column designating the timepoints, in order to pass their order to the LMM model. This example should be relatively clear, however there never is right or wrong with reshaping a dataset – it is all up to your hypothesis and the way you want to encode the information therein. In my experience, in most LMMs it works better but it is not strictly necessary. Btw if this topic interests you I suggest you have a look into my Bayesian modelling tutorial, a fantastic alternative to LMMs which I find more satisfying and intuitive. Hope this helps?
Francisco
LikeLike