

This time I elaborate on a much more specific subject that will mostly concern biologists and geneticists. I will try my best to outline the approach as to ensure non-experts will still have a basic understanding. This tutorial illustrates the power of genome-wide association (GWA) studies by mapping the genetic determinants of cholesterol levels using three Southeast Asian populations.
Historical background
Early since Charles Darwin formulated the theories of natural and sexual selection in the late 1800s, the underlying role of genes, each represented by different alleles (i.e. variants) in different individuals was yet to be elucidated. His younger contemporary fellow Gregor Mendel scratched the surface after identifying the mechanism of trait inheritance, genetic segregation. In the years that followed, the genetic basis of phenotypes (i.e. observable traits) was gradually unraveled by classic geneticists pioneered by Ronald Fisher, who introduced key concepts such as genetic variance. The then emerging concept of genotype (i.e. genetic make-up) required the development of polymorphic genetic markers. In the early days, genotyping was based on determining the allelic composition of loci (loose definition of chromosomal regions), and later of copy number variations (CNVs), short-tandem repeats (STRs) and single nucleotide polymorphisms (SNPs). Humans and many other organisms including plants are diploid (i.e. carry two copies of each chromosome), which implies bi-allelic markers with alleles e.g. A and a distinguish individuals into AA (homozygous dominant), Aa (heterozygous) and aa (homozygous recessive).
The early discovery of the genetic basis for disorders such as sickle cell anemia and haemophilia owes much to their relatively simple genetic architectures – few mutations with high penetrance, thus more easily identified. Understandably then, more complex polygenic diseases that have been around for a long time, such as the neurodegenerative Alzheimer and Parkinson, are still currently under investigation. To characterize such traits, products of the interplay of many genes with small effects, it takes large genetic resources as well as flexible statistical methods, which is no problem in the current era of omics data and high-performance computing.
Genome-wide association studies
Genome-wide association (GWA) studies scan an entire species genome for association between up to millions of SNPs and a given trait of interest. Notably, the trait of interest can be virtually any sort of phenotype ascribed to the population, be it qualitative (e.g. disease status) or quantitative (e.g. height). Essentially, given p SNPs and n samples or individuals, a GWA analysis will fit p independent univariate linear models, each based on n samples, using the genotype of each SNP as predictor of the trait of interest. The significance of association (P-value) in each of the p tests is determined from the coefficient estimate 
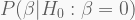
Association mapping vs. linkage mapping
Too often, people cannot tell the difference between association and linkage mapping, or quantitative trait loci (QTL) mapping. Albeit conceptually similar, their are actually opposite in their workings. One of the key differences between the two is that association mapping relies on high-density SNP genotyping of unrelated individuals, whereas linkage mapping relies on the segregation of substantially fewer markers in controlled breeding experiments – unsurprisingly QTL mapping is seldom conducted in humans. Importantly, association mapping gives you point associations in the genome, whereas linkage mapping gives you QTL, chromosomal regions.
The present tutorial covers fundamental aspects to consider when conducting GWA analysis, from the pre-processing of genotype and phenotype data to the interpretation of results. We will use a mixed population of 316 Chinese, Indian and Malay that was recently characterized using high-throughput SNP-chip sequencing, transcriptomics and lipidomics (Saw et al., 2017). More specifically, we will search for associations between the >2.5 million SNP markers and cholesterol levels. Finally, we will explore the vicinity of candidate SNPs using the USCS Genome Browser in order to gain functional insights. The methodology shown here is largely based on the tutorial outlined in Reed et al., 2015. The R scripts and all data can be found in my repository, but you can also download the omics data from here. Please follow the instructions in the repo.
Let’s get started with R
Read data
Let’s first import the PLINK-converted .bed, .fam and .bim Illumina files from each of the three ethnic groups. We will use the function read.plink from the package snpStats and work on the resulting objects throughout the rest of the tutorial. This function reads .bed, .fam and .bim and creates a list of three elements – $genotypes, $fam and $map. The first contains all SNPs determined from all samples, the second contains information about pedigree and sex, and the third contains the genomic coordinates of the SNPs. At this point we have a total of 323 individuals (110 Chinese, 105 Indian and 108 Malay) and 2,527,458 SNPs. Next, we will combine the information from the three SNP datasets and change the Illumina SNP IDs stored in $map$SNP to the more conventional rs IDs, which will allow us to zoom in into the genomic regions that surround the candidate SNPs in the USCS Genome Browser. I prepared a table in advance that establishes the correspondence between the two, so that we can easily switch the IDs.
| library(snpStats) | |
| load("conversionTable.RData") | |
| pathM <- paste("public/Genomics/108Malay_2527458snps", c(".bed", ".bim", ".fam"), sep = "") | |
| SNP_M <- read.plink(pathM[1], pathM[2], pathM[3]) | |
| pathI <- paste("public/Genomics/105Indian_2527458snps", c(".bed", ".bim", ".fam"), sep = "") | |
| SNP_I <- read.plink(pathI[1], pathI[2], pathI[3]) | |
| pathC <- paste("public/Genomics/110Chinese_2527458snps", c(".bed", ".bim", ".fam"), sep = "") | |
| SNP_C <- read.plink(pathC[1], pathC[2], pathC[3]) | |
| # Ensure == number of markers across the three populations | |
| if(ncol(SNP_C$genotypes) != ncol(SNP_I$genotypes)){ | |
| stop("Different number of columns in input files detected. This is not allowed.") | |
| } | |
| if(ncol(SNP_I$genotypes) != ncol(SNP_M$genotypes)){ | |
| stop("Different number of columns in input files detected. This is not allowed.") | |
| } | |
| # Merge the three SNP datasets | |
| SNP <- SNP_M | |
| SNP$genotypes <- rbind(SNP_M$genotypes, SNP_I$genotypes, SNP_C$genotypes) | |
| colnames(SNP$map) <- c("chr", "SNP", "gen.dist", "position", "A1", "A2") # same for all three | |
| SNP$fam<- rbind(SNP_M$fam, SNP_I$fam, SNP_C$fam) | |
| # Rename SNPs present in the conversion table into rs IDs | |
| mappedSNPs <- intersect(SNP$map$SNP, names(conversionTable)) | |
| newIDs <- conversionTable[match(SNP$map$SNP[SNP$map$SNP %in% mappedSNPs], names(conversionTable))] | |
| SNP$map$SNP[rownames(SNP$map) %in% mappedSNPs] <- newIDs |
Next we import and merge the three lipid data sets (stored as .txt) and determine which samples are present in both SNP and lipid data sets. In the following analyses we will use the subset of samples profiled in both platforms, a total of 319. Finally, we create a list genData that stores the merged SNP data ($SNP), one of the three $map since they are all identical ($MAP) and the merged lipid data ($LIP). Finally, let’s save RAM for the subsequent steps and remove all files after saving genData into a .RData file, and the combined $genotypes into a single PLINK file.
| # Load lipid datasets & match SNP-Lipidomics samples | |
| lipidsMalay <- read.delim("public/Lipidomic/117Malay_282lipids.txt", row.names = 1) | |
| lipidsIndian <- read.delim("public/Lipidomic/120Indian_282lipids.txt", row.names = 1) | |
| lipidsChinese <- read.delim("public/Lipidomic/122Chinese_282lipids.txt", row.names = 1) | |
| all(Reduce(intersect, list(colnames(lipidsMalay), | |
| colnames(lipidsIndian), | |
| colnames(lipidsChinese))) == colnames(lipidsMalay)) # TRUE | |
| lip <- rbind(lipidsMalay, lipidsIndian, lipidsChinese) | |
| # Country | |
| country <- sapply(list(SNP_M, SNP_I, SNP_C), function(k){ | |
| nrow(k$genotypes) | |
| }) | |
| origin <- data.frame(sample.id = rownames(SNP$genotypes), | |
| Country = factor(rep(c("M", "I", "C"), country))) | |
| matchingSamples <- intersect(rownames(lip), rownames(SNP$genotypes)) | |
| SNP$genotypes <- SNP$genotypes[matchingSamples,] | |
| lip <- lip[matchingSamples,] | |
| origin <- origin[match(matchingSamples, origin$sample.id),] | |
| # Combine SNP and Lipidomics | |
| genData <- list(SNP = SNP$genotype, MAP = SNP$map, LIP = lip) | |
| # Write processed omics and GDS | |
| save(genData, origin, file = "PhenoGenoMap.RData") | |
| write.plink("convertGDS", snps = SNP$genotypes) | |
| # Clear memory | |
| rm(list = ls()) |
Pre-processing
Let’s reload genData and clean it up. In a nutshell, the pre-processing of the data consists in
- discarding SNPs with call rate < 1 or MAF < 0.1
- discarding samples with call rate < 100%, IBD kinship coefficient > 0.1 or inbreeding coefficient |F| > 0.1
Call rate is the proportion of SNPs (or samples) that were genotyped. For example, a call rate of 0.95 for a particular SNP (sample) means 5% of the values are missing. Because we have so many SNPs, we can afford to have absolutely no missing values in the $SNP matrix by imposing a call rate threshold of 1 for both SNPs and samples. If you want to relax the threshold and tolerate missing values, bear in mind you need to run a whole procedure for imputing those. Reed et al., 2015 describe a PCA-based imputation method that utilizes the 1,000 Genome Project as a proxy, in case you are interested.
Minor-allele frequency (MAF) denotes the proportion of the least common allele for each SNP. Of course, it is harder to detect associations with rare variants and this is why we select against low MAF values. Most GWA studies I have read typically report MAF thresholds of 0.05. Here, I opt for a more stringent 0.1 because again, we have plenty of data and since all this is for illustrative purposes we want to conduct a fast GWA analysis.
| library(snpStats) | |
| library(doParallel) | |
| library(SNPRelate) | |
| library(GenABEL) | |
| library(dplyr) | |
| source("GWASfunction.R") | |
| load("PhenoGenoMap.RData") | |
| # Use SNP call rate of 100%, MAF of 0.1 (very stringent) | |
| maf <- 0.1 | |
| callRate <- 1 | |
| SNPstats <- col.summary(genData$SNP) | |
| maf_call <- with(SNPstats, MAF > maf & Call.rate == callRate) | |
| genData$SNP <- genData$SNP[,maf_call] | |
| genData$MAP <- genData$MAP[maf_call,] | |
| SNPstats <- SNPstats[maf_call,] |
Next, we need to consider samples that exhibit excessive heterozygosity – technically speaking, deviations from the Hardy-Weinberg equilibrium (HWE),
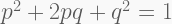
where 
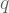


where H and
| # Sample call rate & heterozygosity | |
| callMat <- !is.na(genData$SNP) | |
| Sampstats <- row.summary(genData$SNP) | |
| hetExp <- callMat %*% (2 * SNPstats$MAF * (1 - SNPstats$MAF)) # Hardy-Weinberg heterozygosity (expected) | |
| hetObs <- with(Sampstats, Heterozygosity * (ncol(genData$SNP)) * Call.rate) | |
| Sampstats$hetF <- 1-(hetObs/hetExp) | |
| # Use sample call rate of 100%, het threshold of 0.1 (very stringent) | |
| het <- 0.1 # Set cutoff for inbreeding coefficient; | |
| het_call <- with(Sampstats, abs(hetF) < het & Call.rate == 1) | |
| genData$SNP <- genData$SNP[het_call,] | |
| genData$LIP <- genData$LIP[het_call,] |
Finally, we will investigate relatedness among samples using the kinship coefficient based on identity by descent (IBD). Please note that these functions from the package SNPRelate require GDS files. For this reason we first need to aggregate the .bed, .fam and .bim files from the three populations into convertGDS. The function snpgdsBED2GDS2 creates the GDS necessary for this part of the analysis. To determine the kinship coefficient between pairs of samples we will use a subset of uncorrelated SNPs in order to have unbiased estimates. For this purpose, we will use linkage disequilibrium (LD) as a measure of correlation between markers. LD ranges from 0 to 1, the higher its value the more likely two SNPs co-segregate and therefore correlate. Here, we will utilize the subset of SNPs with LD < 0.2 (p ~ 12,000) to determine the IBD kinship coefficient. It took me about 2 hours to calculate the LD with the function snpgdsLDpruning, so be patient. Next, following Reed et al., 2015, we will exclude all samples with kinship coefficients > 0.1.
| # LD and kinship coeff | |
| ld <- .2 | |
| kin <- .1 | |
| snpgdsBED2GDS(bed.fn = "convertGDS.bed", bim.fn = "convertGDS.bim", | |
| fam.fn = "convertGDS.fam", out.gdsfn = "myGDS", | |
| cvt.chr = "char") | |
| genofile <- snpgdsOpen("myGDS", readonly = F) | |
| gds.ids <- read.gdsn(index.gdsn(genofile, "sample.id")) | |
| gds.ids <- sub("-1", "", gds.ids) | |
| add.gdsn(genofile, "sample.id", gds.ids, replace = T) | |
| geno.sample.ids <- rownames(genData$SNP) | |
| # First filter for LD | |
| snpSUB <- snpgdsLDpruning(genofile, ld.threshold = ld, | |
| sample.id = geno.sample.ids, | |
| snp.id = colnames(genData$SNP)) | |
| snpset.ibd <- unlist(snpSUB, use.names = F) | |
| # And now filter for MoM | |
| ibd <- snpgdsIBDMoM(genofile, kinship = T, | |
| sample.id = geno.sample.ids, | |
| snp.id = snpset.ibd, | |
| num.thread = 1) | |
| ibdcoef <- snpgdsIBDSelection(ibd) | |
| ibdcoef <- ibdcoef[ibdcoef$kinship >= kin,] | |
| # Filter samples out | |
| related.samples <- NULL | |
| while (nrow(ibdcoef) > 0) { | |
| # count the number of occurrences of each and take the top one | |
| sample.counts <- sort(table(c(ibdcoef$ID1, ibdcoef$ID2)), decreasing = T) | |
| rm.sample <- names(sample.counts)[1] | |
| cat("Removing sample", rm.sample, "too closely related to", | |
| sample.counts[1], "other samples.\n") | |
| # remove from ibdcoef and add to list | |
| ibdcoef <- ibdcoef[ibdcoef$ID1 != rm.sample & ibdcoef$ID2 != rm.sample,] | |
| related.samples <- c(as.character(rm.sample), related.samples) | |
| } | |
| genData$SNP <- genData$SNP[!(rownames(genData$SNP) %in% related.samples),] | |
| genData$LIP <- genData$LIP[!(rownames(genData$LIP) %in% related.samples),] |
After pre-processing, we are left with 316 samples (110 Chinese, 100 Indian and 106 Malay) characterised by 795,668 SNP markers and 282 lipid species. Note that your sample size might differ slightly as the LD pruning procedure is stochastic.
Analysis
Principal Component Analysis
Now that we are done with the pre-processing, it might be a good idea to examine the largest sources of variation in the genotype data and look out for outliers or clustering patterns, using Principal Component Analysis (PCA). Because we are working with S4 objects, we will be using the PCA function from SNPRelate, snpgdsPCA. Let’s plot the first two principal components (PCs).
| # PCA | |
| set.seed(100) | |
| pca <- snpgdsPCA(genofile, sample.id = geno.sample.ids, | |
| snp.id = snpset.ibd, num.thread = 1) | |
| pctab <- data.frame(sample.id = pca$sample.id, | |
| PC1 = pca$eigenvect[,1], | |
| PC2 = pca$eigenvect[,2], | |
| stringsAsFactors = F) | |
| # Subset and/or reorder origin accordingly | |
| origin <- origin[match(pca$sample.id, origin$sample.id),] | |
| pcaCol <- rep(rgb(0,0,0,.3), length(pca$sample.id)) # Set black for chinese | |
| pcaCol[origin$Country == "I"] <- rgb(1,0,0,.3) # red for indian | |
| pcaCol[origin$Country == "M"] <- rgb(0,.7,0,.3) # green for malay | |
| png("PCApopulation.png", width = 500, height = 500) | |
| plot(pctab$PC1, pctab$PC2, xlab = "PC1", ylab = "PC2", col = pcaCol, pch = 16) | |
| abline(h = 0, v = 0, lty = 2, col = "grey") | |
| legend("top", legend = c("Chinese", "Indian", "Malay"), col = 1:3, pch = 16, bty = "n") | |
| dev.off() |

As expected, the 795,668 SNP markers clearly delineate the three populations. The results also suggest that Chinese and Malay are closer to each other than to Indian (this observation would be much better addressed with e.g. hierarchical clustering). Also, no obvious outliers are identified with the first two PCs. All good to proceed to GWA.
Genome-Wide Association
Finally, the pièce de résistance. From the set of 282 lipids species I chose cholesterol, one of the most familiar, as the trait of interest. You are completely free to select a different lipid species and proceed. We are going to use the GWA function provided in Reed et al., 2015 with some minor modifications. I recommend you to open the script GWASfunction.R and skim through. This is an excellent, well documented parallelized implementation. Note that the glm function is used to determine the significance of association between each SNP and the trait of interest. In case you are wondering, glm is much more versatile than lm since it conducts Gaussian, Poisson, binomial and multinomial regression / classification, depending on how your trait of interest is distributed (all lipids in the phenotype file are Gaussian). This GWA function will not create a variable, but rather write a .txt summary table listing the coefficient estimate
| # Choose trait for association analysis, use colnames(genData$LIP) for listing | |
| # NOTE: Ignore the first column of genData$LIP (gender) | |
| target <- "Cholesterol" | |
| phenodata <- data.frame("id" = rownames(genData$LIP), | |
| "phenotype" = scale(genData$LIP[,target]), stringsAsFactors = F) | |
| # Conduct GWAS (will take a while) | |
| start <- Sys.time() | |
| GWAA(genodata = genData$SNP, phenodata = phenodata, filename = paste(target, ".txt", sep = "")) | |
| Sys.time() - start # benchmark |
Once finished, we can visualize the results using so-called Manhattan plots. All we need is to load the .txt summary table written in the previous step, add a column with 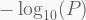
| # Manhattan plot | |
| GWASout <- read.table(paste(target, ".txt", sep = ""), header = T, colClasses = c("character", rep("numeric",4))) | |
| GWASout$type <- rep("typed", nrow(GWASout)) | |
| GWASout$Neg_logP <- -log10(GWASout$p.value) | |
| GWASout <- merge(GWASout, genData$MAP[,c("SNP", "chr", "position")]) | |
| GWASout <- GWASout[order(GWASout$Neg_logP, decreasing = T),] | |
| png(paste(target, ".png", sep = ""), height = 500,width = 1000) | |
| GWAS_Manhattan(GWASout) | |
| dev.off() |
In addition, a full (resp. dashed) line indicate the levels of Bonferroni-adjusted 

We see that a total of four SNPs pass the ‘relaxed’ Bonferroni threshold (none passes the ‘hard’ threshold). These are SNPs rs7527051 (Chr. 1), rs12140539 (co-localized with the first, Chr. 1), rs9509213 (Chr. 13) and rs2250402 (Chr. 15).
Before proceeding with these four hits, it is helpful to constrast the distribution of the resulting P-values against that expected by chance, as to ensure there is no confounding systemic bias. This is easily addressed with a quantile-quantile (Q-Q) plot. As the name suggests, it compares two distributions based on their quantiles. In the present case, we want to contrast the distribution of our t statistics against that obtained by chance. If two distributions are identical in shape, the Q-Q plot will display a 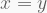
| # QQ plot using GenABEL estlambda function | |
| png(paste(target, "_QQplot.png", sep = ""), width = 500, height = 500) | |
| lambda <- estlambda(GWASout$t.value**2, plot = T, method = "median") | |
| dev.off() |
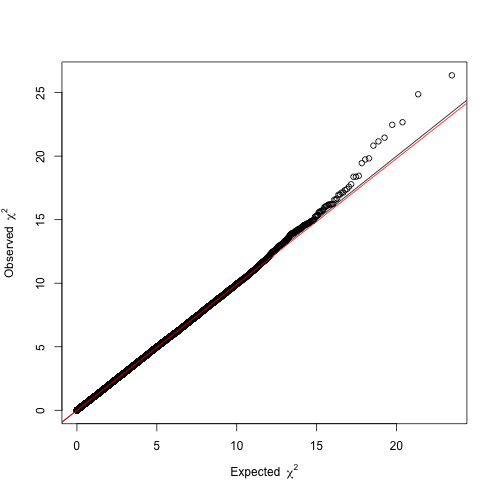
The resulting Q-Q plot clearly depicts a trend line (
Functional insights into candidate markers
Finally, we will try to find the functional relevance of these four candidate SNPs by searching for genes in their vicinity, using the USCS Genome Browser (enter Genome Browser, insert the SNP ID in the text box, enter and zoom out). I found that rs9509213 (Chr. 13) lands right on CRYL1 (crystalline lambda 1, intron sequence), rendering it an interesting candidate for follow-up studies.

The SNP rs9509213 is shown as a black text box in the bottom of the figure, and its coordinates are highlighted by a vertical yellow line. The CRYL1 gene model is shown on top, below the chromosome model (topmost).
Interestingly, there is a recent publication that found ‘POE, HP, and CRYL1 have all been associated with Alzheimer’s Disease, the pathology of which involves lipid and cholesterol pathways.’.
Finally, I would like to remark on the need of experimental validation of gene candidates to validate the resulting SNP-trait associations. In this particular case, one could test the effect of overexpressing or knocking-out the ortholog of CRYL1 in cholesterol levels of mice.
Wrap-up
To sum up, we have covered some of the most fundamental aspects of GWA analysis:
- Pre-processing of genotype data based on call rates, MAF, kinship and heterozygosity
- Investigation of population structure with PCA of the genotype data
- GWA and visualisation with Manhattan plots
- Q-Q plots
- Functional insights into the resulting candidate SNPs
I did not cover many other aspects of GWA, such as fine-mapping using LD plots. Importantly, we did not consider sample size calculation in relation to the expected detectable effect sizes. Also noteworthy, the state-of-the-art of GWA is now almost-fully based on mixed-effect linear models (MLM) that consider kinship and cryptic relatedness as random factors, hence much more powerful that glm. My next post might cover MLM.
I encourage you to choose a different lipid species from the phenotype data, run the analysis and interpret the results.
I would like to thank Saw et al., 2017 for providing unrestricted access to their omics data. If it wasn’t for such conscientious scientists and their transparent research endeavours this whole tutorial would simply not exist. I also reiterate that this tutorial is largely based on the guide outlined in Reed et al., 2015, an excellent reference along with Sikorska et al., 2013.
Have fun and keep your cholesterol in check. Any feedback or suggestions are welcome!
Citation
de Abreu e Lima, F (2017). poissonisfish: Genome-wide association studies in R. From https://poissonisfish.com/2017/10/09/genome-wide-association-studies-in-r/

Wow! Talk about timely! I have an honors student in my undergraduate biostatistics course who is just beginning to write an introductory “GAA for Dummies” tutorial for genome-wide association analysis. This article is a gold mine of information and references. Thanks.
LikeLike
You are welcome! Cheers
LikeLike
Do you know why I can not unzip the iOmics file??? I download and try to use the WinZip but I have got error so far
LikeLike
Hi Mateusz! Which OS are you using? If Mac/Linux, open a terminal, use the ‘cd’ command to find the corresponding directory and type ‘tar xf iOmics_data.tar.gz’. If you are using Windows please use a software other than WinZip and let me know if your issue was solved. Cheers!
LikeLike
While I was running the scripts about “Genome-wide association studies in R” posted at https://www.r-bloggers.com/genome-wide-association-studies-in-r/ , I got the following error message.
> snpgdsBED2GDS(bed.fn = “convertGDS.bed”, fam.fn = “convertGDS.fam”, bim.fn = “convertGDS.bim”, out.gdsfn = “myGDS”, cvt.chr = “char”)
Start snpgdsBED2GDS …
Error in file(filename, “rb”) : cannot open the connection
In addition: Warning message:
In file(filename, “rb”) :
cannot open file ‘convertGDS.bed’: No such file or directory
Can you please tell me what I am doing wrongly.
The code was posted by Francisco Lima, but I could not find his email address.
Thank you!
LikeLike
Hi Ephrem! Did you run the script ‘1-getGDS.R‘?
Please follow the instructions in the repository as the code shown above is not complete! Let me know if this solved your problem, ok? Cheers
LikeLike
Many thanks Francisco.
Everything went smoothly.
Great!
Wanted to know whether you plan also to implement mixed models in GWAS.
Thanks again.
LikeLike
Hi again Ephrem! I am glad it worked. The next post will be out in one or two weeks, it will definitely deal with mixed models using a simple experiment. I can nevertheless add a paragraph or two about the applications in GWAS. Cheers
LikeLike
is it possible to run the snpgdsLDpruning function with more than 1 cpu? there is an argument to set threads but no matter how many I set, the system only utilizes one.
LikeLike
Hi Bo! Is this the error message – “The current version of ‘snpgdsLDpruning()’ does not support multi-threading”? It seems like the current version (at least in the dev-repo) does not support multi-threading (https://github.com/zhengxwen/SNPRelate/blob/master/R/LD.R). Otherwise, should your version be fully operational, examine the code chunk that sets up the parallelisation in that particular function – I have found there are implementations that either work on UNIX/OS or Windows. Hope this helps! Cheers
LikeLike
Hello Francisco, i haven’t been able to install the “GenABEL” package, as it has been discontinued from R. Do you know a way to deal with this problem?
Thanks!
Kind Regards
Nicolás
LikeLiked by 1 person
Hi Nicolas! I hope this link helps you. Take a version from about the time I posted this!
https://support.rstudio.com/hc/en-us/articles/219949047-Installing-older-versions-of-packages
LikeLike
Hi Francisco, I have the same problem cannot install “GenABEL” package even I tried the methods in the link.
I even tried to use old R versions, it’s still not working.
Regards.
LikeLike
Hi Enwu! Take a look here: https://cran.r-project.org/src/contrib/Archive/GenABEL/
Please try installing source, if possible. Something like:
install.packages(packageurl, repos=NULL, type=”source”)
where packageurl points to any tar.gz version in that list. I will investigate the problem a bit further later at night
LikeLike
Hi Francisco! I have a question what kind of new R package would make your life easier when you were doing this analyzation? An embedded genome browser that is using the Genome Browser’s database API? or writing instead of a .txt file, directly reading and drawing a plot?
LikeLike
Hi Doga! I think an interface bridging GWA results and a Genome Browser (more technically gene coordinates that co-localize with selected markers) would be very exciting. To my knowledge this currently still is an open venue, but it suffers from a old standing issue – you have to custom-tailor such interfaces to individual species. Search for tools of that sort based on the human genome (R based or not), if none exist you can take the lead. As for plotting from GWA results (.txt or otherwise), I think there are so many options out there I would not even bother… Hope this helps!
LikeLike
Hi Francisco! Amazing post! I’m working with GWAS data, I have the location (chromosome and genome) of each SNP. I already sucess plotting the SNPs just using the chromosome locations, but dont how to plot then using the genome as well (1A).
Hope I’m clear:) Thanks in advance!
LikeLiked by 1 person
Thanks Ale! If I understood correctly, for each SNP you have the coordinates that map into the corresponding chromosome (that is, you have both position and chromosome). In that case, you can produce a Manhattan plot by ordering all SNPs AND chromosomes. In terms of code, you could try encoding new coordinates that extend to all the ordered chromosomes. For example, the first position in Chr. 4 would be the last position in Chr. 3 plus 1bp. Overall, these new coordinates would be the x-axis of your figure. Does this help?
LikeLike
Hi Francisco,
I really liked your post.
I tried to perform the very same analysis, but I don’t know where to find the conversionTable.RData image. Nor do I know how to obtain the PLINK-converted .bed, .fam and .bim Illumina files from each of the three ethnic groups you mention.
Sincerely
Hervé
LikeLike
Hi Hervé, how are you? Please check the response to Ephram above!
LikeLike
Hi Francisco,
Yeah you are right I really should have paid more attention.
I now have dowloaded the files and I am trying to follow all the described steps.
Hope I will be able to run the GenABEL part, as I run the snpStats part with R 5.1, which appears to be non compatible with my previous GenABEL library (compiled for R 3.3.3).
I will let you know if I run into difficulties I cannot solve despite all the feedback.
Sincerely,
Hervé
LikeLike
Hi again Hervé. Please do let me know if it does not work! Cheers
LikeLike
Thanks for this article, it is a very good introduction to that subject.
LikeLike
Merci Francois!
LikeLike
UPDATE / ERRATUM
Fix for Linux/macOS installation of GenABEL
install.packages(“GenABEL.data”, repos=”http://R-Forge.R-project.org”)
packageurl <- "https://cran.r-project.org/src/contrib/Archive/GenABEL/GenABEL_1.8-0.tar.gz"
install.packages(packageurl, repos=NULL)
Fix for dplyr `arrange` and `count` in `while` loop
while (nrow(ibdcoef) > 0) {
# count the number of occurrences of each and take the top one
sample.counts <- sort(table(c(ibdcoef$ID1, ibdcoef$ID2)), decreasing = T)
rm.sample <- names(sample.counts)[1]
cat("Removing sample", rm.sample, "too closely related to", sample.counts[1], "other samples.\n")
# remove from ibdcoef and add to list
ibdcoef <- ibdcoef[ibdcoef$ID1 != rm.sample & ibdcoef$ID2 != rm.sample,]
related.samples <- c(as.character(rm.sample), related.samples)
}
LikeLike
Hi Francisco,
I thought the Package ‘GenABEL’ was removed from the CRAN repository due to some non-compliances.
have a nice weekend.
LikeLike
Hi Ephrem,
Yes, it was removed from CRAN presumably because it was no longer supported. You can install it as per above, based on archived releases.
LikeLike
Hi Francisco,
Your post is amazing. I have a question regarding using my own database as it comes from axiom Arachis microarray I haven’t got the PLINK file. I only have genotypes for my samples and the chromosome and position for each SNP. Is it possible to run this analysis following your tutorial? Thank you very much in advance
LikeLike
Hi frandeblas, thanks for the kind words. There certainly are tons of tools you can use to convert files over a wide range of formats including PLINK. What format are your files?
Best,
Francisco
LikeLike
Thank you for the quick response! I have .txt or .csv files for the phenotype, genotypes and SNPs information.
Thanks a lot!
LikeLike
Can you use the PLINK binary with the option –recode ? Check info under:
http://www.cog-genomics.org/plink/1.9/data#make_bed
http://zzz.bwh.harvard.edu/plink/dataman.shtml
LikeLike
Dear Francisco,
thank you for your post.
Unfortunatelly, the GWAA function doesn’t work to me…
Please, see the error:
Error in { : task 1 failed – “task 1 failed – “indice fuori limite””
4.
stop(simpleError(msg, call = expr))
3.
e$fun(obj, substitute(ex), parent.frame(), e$data)
2.
foreach(part = 1:nSplits) %do% {
genoNum <- as(genodata[, snp.start[part]:snp.stop[part]],
"numeric")
if (isTRUE(flip)) … at C:\Users\Gabry\Desktop\SNP UniMi Gabriella_2019\QTL_L22A\GWAS_RStudio\GWAA.R#78
1.
GWAA(genodata = genData$SNP, phenodata = phenodata,
filename = paste(target, ".txt", sep = ""))
LikeLike
Hi Gabriella, are you using the scripts from the GitHub repository in the correct order? Which part / script failed?
LikeLike
Thanks for the wonderful article. I have a naive question about PCA: in PCA plot, we see a clear clustering pattern with respect to three countries; is it helpful to incorporate countries as covariate in regression to control possible confounding?
Thanks in advance
LikeLike
Hi Boxi, thanks for your feedback, that is a great question! You could pass the country covariate to the model; however, a more rigorous and unbiased approach to address this problem of so-called population structure is to pass the first N principal components from the PCA to the GLM model in GWASfunction.R, line 60 (see the repo link above), which should ideally only keep associations that are consistent across the entire sample, regardless of country of origin and additional sociodemographic or unknown factors. A good outline of mixed-model GWAS approaches that control for both population structure and familial relatedness can be found in this seminal paper
https://www.nature.com/articles/ng1702 . The corresponding pdf is freely available from https://www.researchgate.net/publication/281071967_A_unified_mixed-model_method_for_association_mapping_that_accounts_for_multiple_levels_of_relatedness . Finally, if you are interested I wrote a post about linear mixed-effect models in R, alas not in the scope of GWAS: https://poissonisfish.com/2017/12/11/linear-mixed-effect-models-in-r/ . I hope this information helps. Happy New Year!
LikeLike
Thank you Francisco, they are really helpful. I’ll play with GWASfunction and read about LMM and population structure issue. Happy new year:D
LikeLike
Hi Francisco, What is the SNP encoding scheme that has been used? And at which step encoding is done? I saw that the SNP columns contain values 1,2,3. What is the basis for this system of encoding? I have read about 0,1,2 encoding scheme which is based on the minor allele count.
LikeLike
Hi, indeed you do normally observe a 0, 1, 2 encoding for biallelic loci as you pointed out – that is the expected output from PLINK (https://zzz.bwh.harvard.edu/plink/dataman.shtml) which is used in the tutorial. Can you paste here any such odd example? Maybe you can go back to the original file and look into those variants / alleles and deduce how they are encoded? Greetings
LikeLike
Hello Francisco, While I was running the scripts about “Genome-wide association studies in R” posted at https://www.r-bloggers.com/genome-wide-association-studies-in-r/ , I got the following error message
Can you please tell me what I am doing wrongly.
LikeLike
Hi Min, thanks for the feedback. Did you use the preprocessing script? Did you effectively write the PLINK files? https://github.com/monogenea/GWAStutorial/blob/master/1-preProc.R
LikeLike
Thank you Francisco. I have checked the script again and it is working! Thank you for the quick response!
LikeLike
Hi Francisco.
Thank you for the tutorial, it is amazing!!
Can you please tell me if it is possible to do an LD heatmap with the provided data?
LikeLike
Hi Devi thank you for your feedback! Yes, I think this is possible – you can perhaps search for such functionality from snpStats or similar, compatible packages
LikeLike
Hi Francisco,
Greetings, firstly I express my appreciation for the elaborate GWAS analysis scripting provided here.
Sadly, I have a difficulty loading in the conversionTable.Rdata, (conversionTable <- read.delim(“conversionTable.txt”, sep = “\t”, stringsAsFactors = F)
load(conversionTable)) I followed the script 1getGDS.R you suggested to Ephrhem and Hervé above. I got an error Error in file(file, “rt”) : cannot open the connection
In addition: Warning message:
In file(file, “rt”) :
cannot open file ‘conversionTable.txt’: No such file or directory
is the conversionTable you prepared in an updated thread? what do you suggest?
Thanks
LikeLike
Hi Ade, thanks a lot for your feedback! You should simply load conversionTable.Rdata:
load(“conversionTable.RData”)
This will load an object to memory named “conversionTable”. Can you please try again?
LikeLike
Many thanks for the swift response Francisco, Got it.
LikeLike